
Our work focuses on understanding how RAS signaling operates in normal cells and how these mechanisms become dysregulated in disease.
RAS proteins act as molecular switches that integrate extracellular cues to control cell growth, survival, and development. When this regulation fails, RAS signaling becomes hyperactive, driving aggressive and treatment-resistant cancers as well as developmental disorders.
We define how RAS signaling is regulated at the molecular level, how it engages key effector pathways, and how these processes are altered in cancer and developmental disease.
That’s what we do.
RAS signaling activation
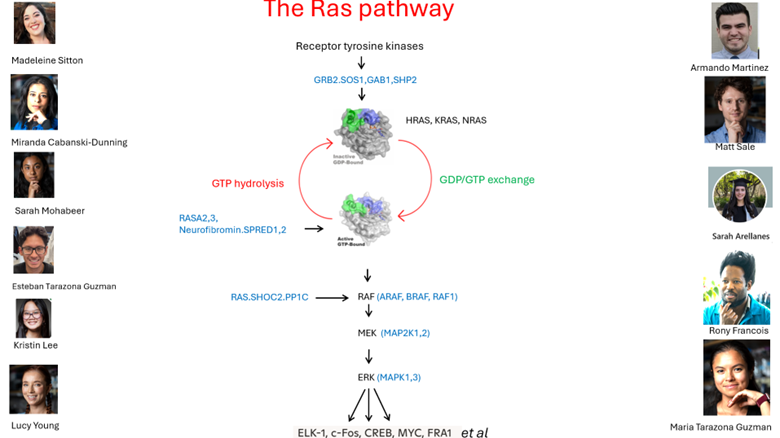
RAS activation is initiated downstream of receptor tyrosine kinases such as EGFR through GEF-mediated GDP–GTP exchange. Adaptor complexes recruit GEFs to the plasma membrane, enabling spatial and temporal control of signaling. Our work investigates how these assemblies regulate pathway output across cellular and disease contexts.
RAS regulation by GAPs
GAP proteins are essential negative regulators of RAS signaling. Seminal work by Frank McCormick and colleagues identified p120GAP and NF1, establishing GAP loss as a major driver of sustained RAS activation in cancer and developmental syndromes. We continue to study how GAP regulation, localization, and context shape signaling.
RAF and MAPK pathway control
RAF activation requires RAS binding, membrane recruitment, and relief of autoinhibition. Our lab has contributed to defining key regulatory mechanisms, including the MRAS–PP1–SHOC2 complex. Current work focuses on how RAF activity is dynamically tuned and how these controls are altered in disease.
RAS–PI3K effector signaling
RAS can directly engage the p110α catalytic subunit of PI3K to promote AKT signaling in specific contexts. We investigate how RAS–PI3K interactions contribute to oncogenic signaling, therapeutic response, and drug resistance.